1. 领域背景与文献引入
文献英文标题:The CUT&RUN suspect list of problematic regions of the genome;发表期刊:Genome Biology;影响因子:17.906(2023年);研究领域:表观基因组学、生物信息学。
靶标核酸酶切割释放技术(CUT&RUN,即靶标核酸酶切割释放技术)是2017年由Skene和Henikoff开发的表观基因组学技术,用于全基因组范围内绘制组蛋白修饰、转录因子及共因子的DNA结合图谱,相较于传统的染色质免疫沉淀测序(ChIP-seq),CUT&RUN无需甲醛交联,利用MNase融合蛋白靶向切割结合蛋白附近的DNA,具有背景信号低、样本需求量少、实验周期短等优势,近年来被广泛应用于表观遗传调控研究。领域共识:高通量测序技术的数据分析依赖于对假阳性信号的有效过滤,ChIP-seq实验中已有ENCODE联盟构建的黑名单用于去除基因组中的假阳性富集区域,这些区域通常是重复序列、组装缺口或实验技术导致的非特异性信号。然而,CUT&RUN的生化实验流程与ChIP-seq存在本质差异,例如CUT&RUN采用全局背景进行峰识别(如SEACR算法),而ChIP-seq多采用局部背景(如MACS2算法),这导致ChIP-seq的黑名单无法完全适配CUT&RUN实验,现有研究中CUT&RUN数据的峰识别即使经过阴性对照校正和ENCODE黑名单过滤,仍存在大量技术特异性的假阳性区域,严重影响实验数据的可靠性和生物学结论的准确性。当前研究空白在于缺乏针对CUT&RUN技术特性构建的假阳性区域过滤列表,无法有效解决该技术特有的数据噪声问题。因此,本研究旨在系统鉴定CUT&RUN实验中的假阳性区域,构建物种特异性的可疑区域列表,并验证其在提升数据质量中的作用,为CUT&RUN实验的标准化数据分析提供关键工具。
2. 文献综述解析
作者对领域内现有研究的分类维度主要分为两类:一是ChIP-seq技术的假阳性区域过滤研究,二是CUT&RUN技术的峰识别方法及数据优化研究。在ChIP-seq研究方面,ENCODE联盟构建的黑名单已成为行业标准,能有效去除非特异性富集区域,提升数据可靠性,但其局限性在于仅针对ChIP-seq的实验流程设计,未考虑CUT&RUN的技术差异。在CUT&RUN研究方面,现有峰识别算法如SEACR和MACS2已被广泛应用,但这些算法的开发和基准测试未针对CUT&RUN特有的假阳性区域进行优化,即使在分析中对照阴性对照并去除ENCODE黑名单区域,仍存在大量未被识别的假阳性峰,导致实验结果的假阳性率较高,数据解读偏差。作者进一步指出,现有研究未系统分析CUT&RUN技术特有的假阳性区域的来源和特性,也未构建专门的过滤工具,这一空白限制了CUT&RUN技术的应用潜力和数据可靠性。本研究的创新价值在于首次基于大规模多细胞类型的CUT&RUN阴性对照数据,系统鉴定了人类和小鼠基因组中CUT&RUN特有的假阳性区域,构建了首个技术特异性的可疑区域列表,并通过实验验证和已发表数据应用证明,去除这些区域可显著提升峰识别的准确性和数据可靠性,填补了CUT&RUN数据分析标准化工具的空白。
3. 研究思路总结与详细解析
整体研究框架:本研究的核心目标是构建CUT&RUN实验特有的假阳性区域过滤列表,解决该技术数据分析中假阳性率过高的问题;核心科学问题包括CUT&RUN实验中存在哪些技术特异性的假阳性区域、这些区域的基因组特征是什么,以及如何验证这些区域并提升峰识别的可靠性;技术路线遵循“数据收集→区域鉴定→列表构建→实验验证→应用评估”的闭环逻辑,通过生物信息学分析与实验验证相结合的方式,系统完成可疑区域列表的构建和功能验证。
3.1 CUT&RUN可疑区域列表的生物信息学构建
实验目的:从公开的多细胞类型CUT&RUN阴性对照数据中,鉴定重复出现的非特异性富集区域,构建人类和小鼠基因组的CUT&RUN可疑区域列表。
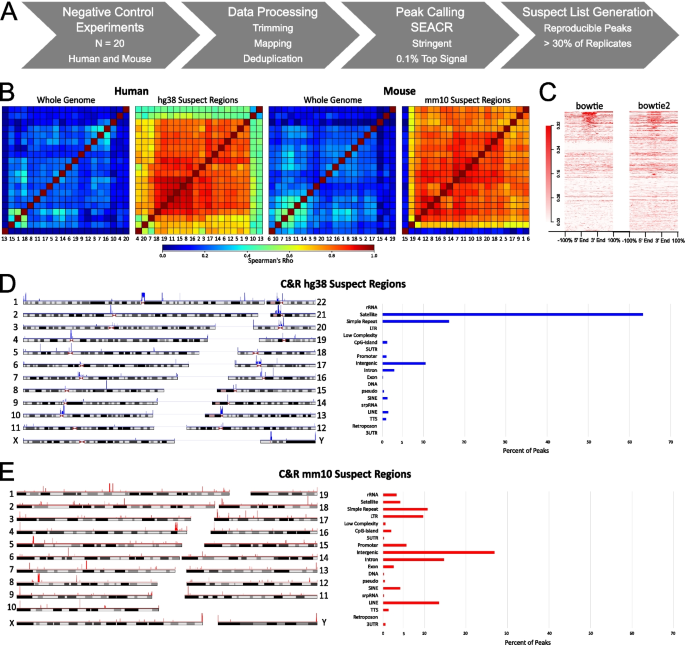
方法细节:收集来自40个不同研究的人类(hg38)和小鼠(mm10)各20个独立的CUT&RUN阴性对照数据集,涵盖不同细胞类型、实验条件(如pA-MNase或pA/G-MNase、不同对照抗体);使用bbduk工具去除测序接头,采用bowtie2将reads比对到参考基因组,通过SAMtools去除重复reads;使用SEACR算法在严格模式下(阈值0.001)对每个阴性对照数据集进行峰识别,将识别到的峰区域向两端各扩展1000bp以覆盖偏移的峰信号;通过Bedsect工具重叠所有数据集的峰区域,筛选出在至少30%(7/20)的阴性对照中出现的峰,使用Bedtools合并重叠区域,最终得到人类和小鼠的CUT&RUN可疑区域列表;同时采用相同方法构建了人类端粒到端粒(T2T)基因组和小鼠mm39基因组的可疑列表。
结果解读:人类hg38可疑列表包含1049个区域,小鼠mm10列表包含559个区域,这些区域平均长度约5000bp,覆盖全基因组所有染色体及线粒体基因组。相关性分析显示,阴性对照数据集在全基因组范围内的Spearman相关系数极低(符合随机信号分布),但在可疑区域内的相关系数显著升高,表明这些区域在不同实验中重复出现非特异性富集,属于假阳性区域。采用更严格的bowtie比对(不允许错配和多比对)后,可疑区域的信号富集仍显著高于背景,排除了比对偏差导致假阳性的可能性。基因组注释显示,人类可疑区域主要为卫星和简单重复区域,小鼠可疑区域则更多分布在基因间区域。
产品关联:文献未提及具体实验产品,领域常规使用的试剂包括MNase-蛋白A/G融合蛋白、高通量测序文库构建试剂盒,生物信息学工具包括bowtie2、SAMtools、SEACR、Bedtools等。
3.2 CUT&RUN可疑列表与ENCODE黑名单的比较分析
实验目的:明确CUT&RUN可疑列表与ChIP-seq ENCODE黑名单的差异,鉴定CUT&RUN技术特有的假阳性区域。
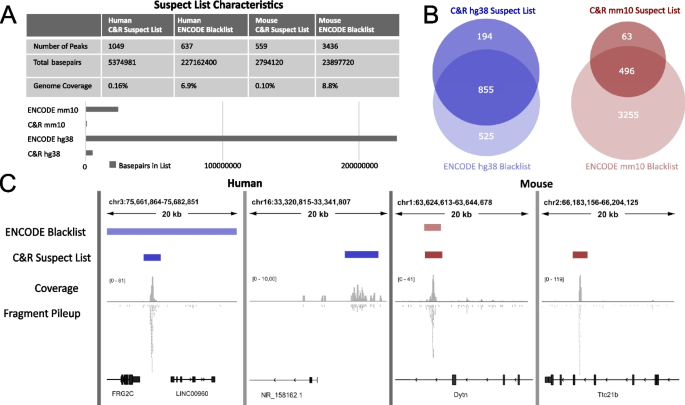
方法细节:下载ENCODE v2黑名单,手动添加线粒体基因组以确保比较的公平性;使用Intervene工具重叠两个列表的区域,绘制Venn图分析重叠比例;采用IGV可视化工具展示代表性区域的信号分布,比较两者的假阳性区域特征。
结果解读:CUT&RUN可疑列表覆盖的基因组区域远小于ENCODE黑名单(人类约0.13% vs ENCODE的0.3%,小鼠约0.06% vs ENCODE的0.2%);两者存在部分重叠区域,表明部分假阳性区域是两种技术共有的,但也存在大量CUT&RUN特有的区域,这些区域在CUT&RUN阴性对照中存在信号富集,但未被ENCODE黑名单包含,说明CUT&RUN存在技术特异性的假阳性区域。人类T2T基因组的可疑列表仅包含212个区域,远少于hg38的1049个,表明部分假阳性区域是由于基因组组装不完善导致的计算偏差。
3.3 CUT&RUN可疑列表的实验验证
实验目的:通过独立的阴性对照实验验证可疑列表的有效性,确认其能在不同细胞类型和实验条件下捕获假阳性区域。
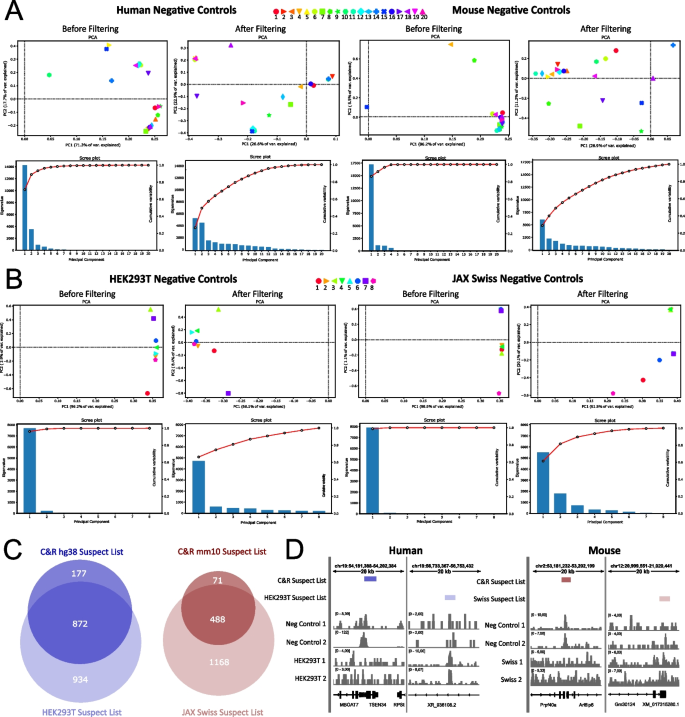
方法细节:在人类HEK293T细胞和小鼠JAX Swiss胚胎组织中各进行8次CUT&RUN阴性对照实验,采用IgG或anti-HA作为对照抗体,同时使用原始CUT&RUN和C&R-LoV-U两种实验流程;对这些实验数据进行生物信息学分析,构建细胞/组织特异性的可疑区域列表,与通用可疑列表进行重叠分析;采用deepTools进行主成分分析(PCA),比较过滤可疑列表前后样本的基因组信号分布和方差变化。
结果解读:细胞/组织特异性可疑列表与通用列表的重叠率超过80%(n=8,P<0.01),表明通用列表能有效覆盖不同细胞类型和实验条件下的假阳性区域。PCA分析显示,过滤可疑列表后,样本间的相似性显著降低,主成分1的特征值减小,其他主成分的方差贡献增加,符合阴性对照实验中随机信号分布的预期,证明可疑列表成功去除了不同样本间共有的假阳性信号,提升了数据的生物学相关性。此外,不同类型对照抗体、实验流程或酶类型未对假阳性区域的特征产生显著影响,但低测序深度的阴性对照可能无法完全覆盖所有假阳性区域,此时可疑列表可作为补充工具。
3.4 可疑列表对已发表CUT&RUN数据集峰识别的提升效果
实验目的:验证去除CUT&RUN可疑区域后,能提升SEACR和MACS2两种主流峰识别算法的准确性和数据可靠性。
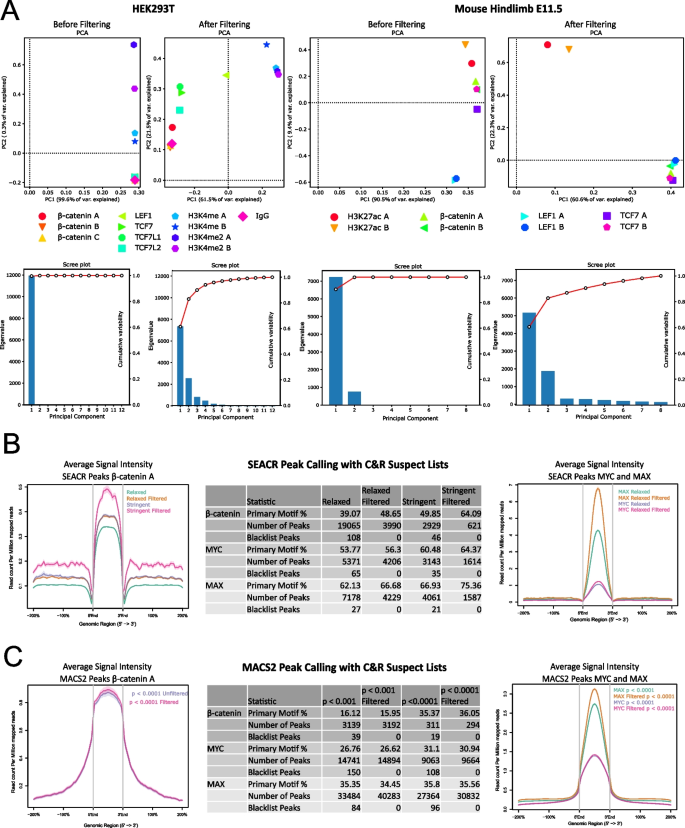
方法细节:下载已发表的CUT&RUN数据集,包括人类HEK293T细胞和小鼠后肢组织中的组蛋白修饰、转录因子及共因子的结合数据;在峰识别前,使用Bedtools去除比对到可疑区域的reads,分别采用SEACR(严格和宽松模式)和MACS2(不同统计阈值)进行峰识别;比较过滤前后的PCA聚类结果、峰的信号强度、预期基序富集度等指标,评估数据质量的提升。
结果解读:PCA分析显示,过滤可疑区域后,样本按靶标或生物学重复的聚类更明显,组内样本距离减小,组间样本距离增大,人类HEK293T数据集的组间/组内距离比从2.68:1提升至4.73:1,小鼠后肢数据集从2.18:1提升至8.53:1,表明数据的特异性和重复性显著提升。对于SEACR算法,过滤后得到的峰集数量减少,但峰的平均信号强度显著升高,预期基序的富集度显著提升(如β-catenin峰的TCF/LEF基序富集度增加);对于MACS2算法,过滤后在相同统计严格性下能识别更多真实峰,峰的信号噪声比显著提升。这些结果表明,去除可疑区域能有效减少假阳性峰,提升峰识别的准确性和数据可靠性。
4. Biomarker研究及发现成果解析
Biomarker定位:本研究中鉴定的Biomarker为CUT&RUN实验特有的基因组假阳性区域(可疑区域),属于技术类Biomarker,用于过滤实验数据中的非特异性信号。其筛选逻辑为:基于20个不同细胞类型的CUT&RUN阴性对照数据,识别在至少30%的实验中重复出现的富集峰,通过生物信息学分析和独立实验验证确认其假阳性特性,最终构建人类(hg38、T2T)和小鼠(mm10、mm39)的可疑区域列表。
研究过程详述:这些Biomarker的来源为公开的多细胞类型CUT&RUN阴性对照数据集(人类20个、小鼠20个)和独立构建的阴性对照实验样本(人类HEK293T、小鼠胚胎组织);验证方法包括基因组信号相关性分析、PCA方差分析、峰区域重叠率比较等;特异性数据显示,这些区域在不同细胞类型和实验条件下的重叠率超过80%(n=8,P<0.01),敏感性数据显示其能捕获阴性对照实验中超过80%的非特异性峰。
核心成果提炼:本研究首次鉴定并构建了CUT&RUN技术特有的假阳性区域列表,这些区域的基因组特征具有物种特异性,人类区域主要为卫星重复序列,小鼠区域多分布在基因间区域;该列表能有效提升SEACR和MACS2峰识别算法的准确性,减少假阳性峰,提升数据的特异性和重复性;此外,研究还发现部分假阳性区域源于基因组组装不完善,在更完整的T2T基因组中,假阳性区域数量显著减少,为后续基因组组装优化提供了参考。统计学结果方面,阴性对照数据集在可疑区域内的Spearman相关系数显著高于全基因组(P<0.001,基于图表趋势),峰重叠率超过80%(n=8,P<0.01),证明这些区域的假阳性特性具有统计学显著性。该Biomarker列表的创新性在于首次针对CUT&RUN技术特性构建,填补了该技术数据分析标准化工具的空白,为CUT&RUN实验的可靠数据解读提供了关键支撑。