1. 领域背景与文献引入
文献英文标题:Coverage and error models of protein-protein interaction data by directed graph analysis;发表期刊:Genome Biology;影响因子:未公开;研究领域:蛋白质相互作用组学、计算生物学
在2007年之前的十年间,酵母双杂交(Y2H)和亲和纯化-质谱(AP-MS)等高通量技术的规模化应用,产生了大量细胞系统中的蛋白质相互作用数据,为分子和计算生物学家全面解析细胞系统及其功能模块提供了可能。然而,现有研究普遍指出高通量蛋白质相互作用数据存在明显的噪音和低质量问题,不同数据集间存在显著差异、高错误率、重叠度低甚至实验结论矛盾等情况,如何准确解读和整合这些大规模数据成为计算生物学面临的重大挑战。
现有研究大多仅基于阳性检测结果推断蛋白质间的相互作用,即阳性结果表示存在相互作用,阴性或无结果则表示不存在,这种方法未充分利用测试集范围、双向验证的互惠性、相互作用类型等关键信息,导致对数据误差的评估和分析严重不足,无法准确估计真实的蛋白质相互作用组及其功能模块。因此,本研究旨在通过有向图模型和统计误差模型,系统分析酿酒酵母所有已发表大规模蛋白质相互作用数据集的误差统计特征,为准确估计蛋白质相互作用组及其模块提供必要前提。
2. 文献综述解析
作者对现有研究的分类维度主要为技术类型(Y2H和AP-MS)和数据质量问题(系统误差、随机误差、覆盖偏差),核心评述逻辑是从数据产生、数据解读到误差分析的全链条,指出现有研究在误差评估和数据利用上的关键缺陷。
现有研究的关键结论是高通量技术已产生海量蛋白质相互作用数据,为细胞系统功能解析提供了基础,但数据本身存在高噪音、高错误率的问题,不同数据集间的一致性差,严重影响了数据的可靠性;技术方法的优势在于Y2H和AP-MS能够实现大规模、高通量的蛋白质相互作用筛选,其中Y2H可有效检测蛋白质间的直接物理相互作用,AP-MS可鉴定蛋白质复合物的组成成员;局限性则体现在多数研究仅报告阳性检测结果,未明确区分未测试的蛋白质对与阴性检测结果,无法准确判断无结果的生物学意义;同时未利用双向测试的互惠性信息识别系统误差,对数据中的系统偏差和随机噪音的分析不足,导致对相互作用数据的解读存在较大偏差,无法为后续的系统生物学建模提供可靠的数据基础。
本研究的创新价值在于首次将有向图模型应用于蛋白质相互作用数据的表征,明确区分测试与未测试的相互作用对,利用双向测试的互惠性特征识别存在系统偏差的蛋白质,建立多项误差模型量化随机误差率,解决了现有研究中误差分析不全面、数据利用不充分的核心问题。通过与现有研究的对比,本研究不仅提供了一套系统的蛋白质相互作用数据质量评估方法,还为后续的相互作用组估计和系统生物学建模提供了关键的误差统计信息,填补了领域内对高通量相互作用数据误差建模的空白。
3. 研究思路总结与详细解析
本研究的整体框架为:以酿酒酵母的12个公开高通量蛋白质相互作用数据集为研究对象,核心目标是系统评估数据的覆盖度、系统误差和随机误差,为准确估计蛋白质相互作用组提供前提;核心科学问题是如何通过有向图建模和统计分析,量化高通量相互作用数据中的各类误差;技术路线遵循“数据收集→模型构建→覆盖度分析→系统误差识别→随机误差估计→结论应用”的闭环逻辑,通过多维度的统计分析全面解析数据的误差特征。
3.1 数据集收集与有向图建模
实验目的:构建标准化的蛋白质相互作用数据表征模型,明确数据中的测试与未测试相互作用对,为后续的误差分析奠定基础。
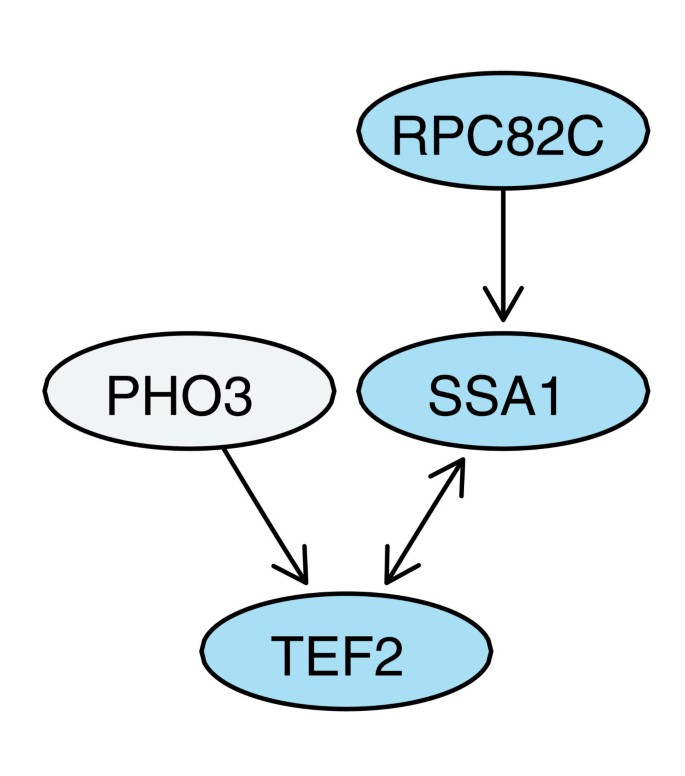
方法细节:收集12个酿酒酵母的公开高通量蛋白质相互作用数据集,其中7个采用Y2H技术,5个采用AP-MS技术,所有数据集均保留诱饵-猎物的方向性信息且猎物为全基因组范围;采用有向图模型表征每个数据集,蛋白质对应图的节点,有向边(b,p)表示诱饵b检测到猎物p,去除自环(同源二聚体相互作用);定义可行诱饵为至少与一个猎物存在相互作用的节点,可行猎物为至少被一个诱饵检测到的节点,双向测试对为同时属于可行诱饵和可行猎物的蛋白质对。
结果解读:通过有向图建模,成功区分了数据集中的测试与未测试相互作用对,明确了双向测试的蛋白质对范围,为后续的误差分析提供了清晰的结构基础;如图1所示,该图展示了Krogan等数据集中4个蛋白质的有向图结构,双向边表示双向均检测到相互作用,单向边表示仅单向检测到,孤立节点表示未被测试,直观呈现了有向图模型的表征效果。
产品关联:文献未提及具体实验产品,领域常规使用的试剂/仪器包括酵母双杂交试剂盒、质谱仪、生物信息学分析软件(如R、Bioconductor)。
3.2 相互作用组覆盖度分析
实验目的:评估高通量实验对酿酒酵母蛋白质相互作用组的覆盖偏差,明确不同技术的覆盖局限性。
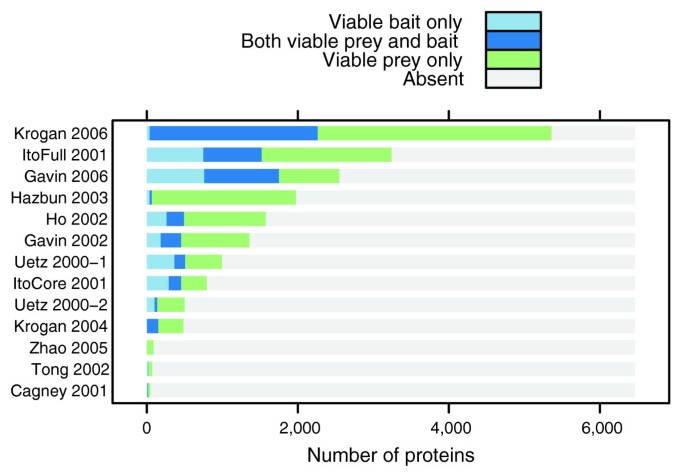
方法细节:将酿酒酵母蛋白质组分为仅可行诱饵(VB)、仅可行猎物(VP)、双向可行(VBP)和存在系统偏差的蛋白质四个子集;采用条件超几何检验分析各子集在基因本体(GO)细胞组分注释中的富集或缺失情况,设定P值阈值为0.01,判断是否存在统计学意义的覆盖偏差。
结果解读:分析结果显示,细胞核(主要为Y2H数据)、细胞质、蛋白质复合物等GO类别的蛋白质在可行蛋白质子集中过度富集,而线粒体、核糖体(Y2H数据)、液泡(AP-MS数据)、膜整合蛋白等类别则过度缺失,说明Y2H和AP-MS技术均存在明显的覆盖偏差,受限于技术本身的实验条件(如Y2H的检测发生在细胞核内,对核内蛋白质更有效;AP-MS难以有效检测膜结合蛋白);如图2所示,该柱状图展示了各数据集的蛋白质采样比例,除Krogan等的数据集外,其余11个数据集均有大量蛋白质未参与任何阳性相互作用,进一步证实了实验的覆盖局限性。
产品关联:文献未提及具体实验产品,领域常规使用的试剂/仪器包括GO注释数据库、统计分析软件。
3.3 系统误差识别与关联分析
实验目的:识别数据集中存在系统偏差的蛋白质,解析系统偏差与蛋白质生物学属性的关联机制。
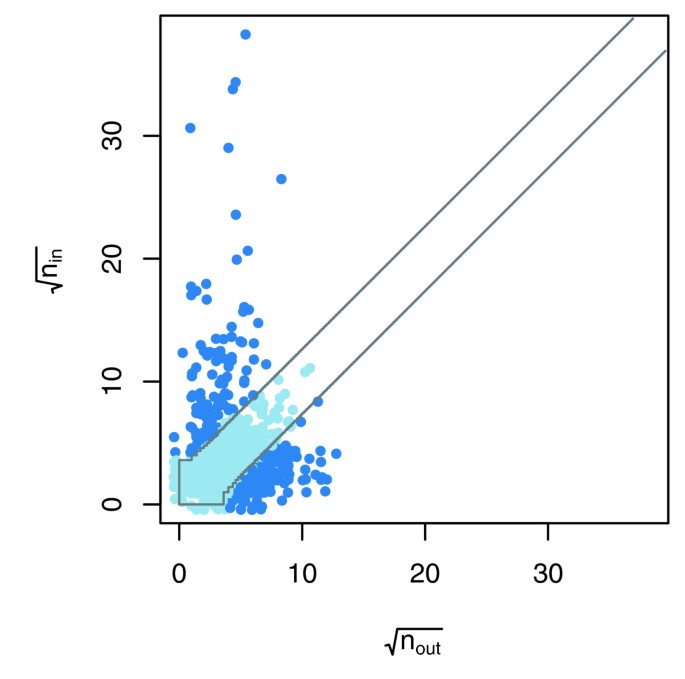
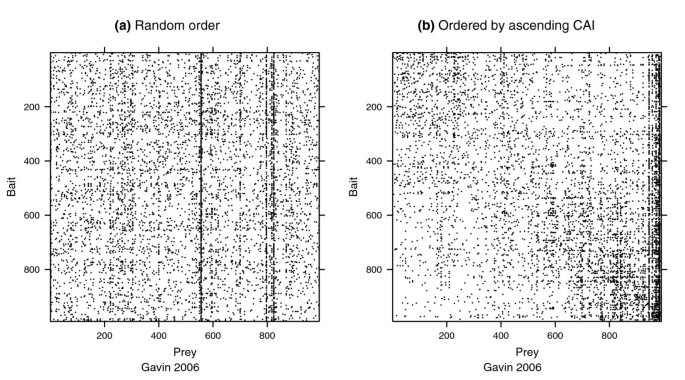
方法细节:针对双向测试的VBP蛋白质,采用二项误差模型,假设未互惠的入度(作为猎物被检测到的次数)和出度(作为诱饵检测到猎物的次数)概率相等,通过双侧二项检验(P<0.01)识别存在系统偏差的蛋白质;采用逻辑回归模型分析存在系统偏差的蛋白质与31种蛋白质属性(如密码子适应指数CAI、蛋白质丰度)的关联;通过邻接矩阵可视化CAI与蛋白质未互惠入度的关联,将矩阵的行和列按CAI升序排列,观察相互作用的分布模式。
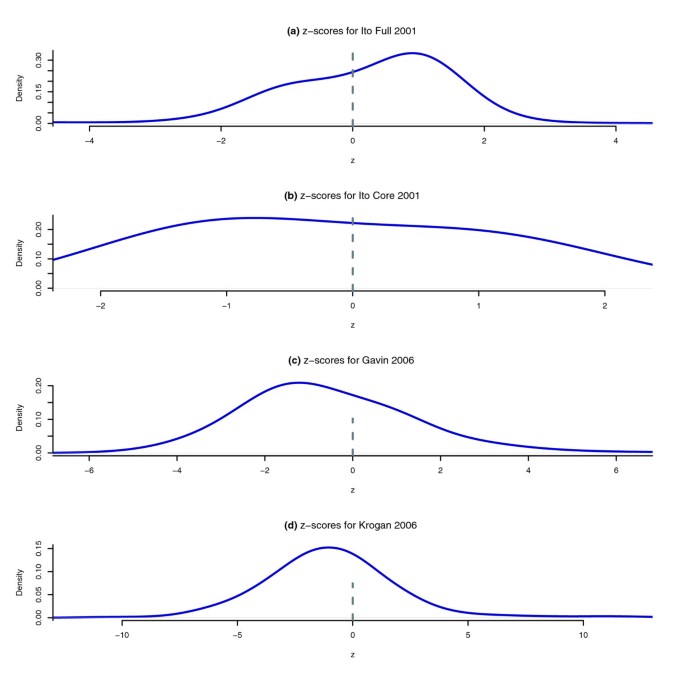
结果解读:统计结果显示,Y2H实验和小规模AP-MS实验中,存在系统偏差的VBP蛋白质比例较低(<3%),而两个大规模AP-MS实验中该比例较高(>14%);在AP-MS数据中,密码子适应指数(CAI)和蛋白质丰度与VBP蛋白质的未互惠入度显著相关,高CAI的蛋白质更容易被作为猎物检测到,导致入度偏高;如图3所示,该散点图展示了Krogan等数据集中二项检验的结果,偏离对角线的蛋白质表示未互惠入度和出度存在显著差异,即存在系统偏差;如图4所示,Gavin等数据集的邻接矩阵按CAI排序后出现明显的垂直暗带,说明高CAI的蛋白质作为猎物被检测到的频率更高;如图5所示,入度z分数的密度分布显示,Ito-Full数据集的z分数均值显著偏高,与数据中存在大量自激活诱饵有关,而Ito-Core数据集通过筛选消除了该效应,z分数分布更接近正态分布。
产品关联:文献未提及具体实验产品,领域常规使用的试剂/仪器包括统计分析软件、蛋白质丰度数据库、基因注释数据库。
3.4 随机误差率的模型估计
实验目的:建立随机误差统计模型,量化数据中的假阳性(FP)和假阴性(FN)率,评估数据的随机噪音水平。
方法细节:基于双向测试的VBP蛋白质对,定义真实相互作用对集合I和非相互作用对集合I^C,建立多项误差模型,通过矩估计法,利用观察到的互惠相互作用数(x₁)、互惠非相互作用数(x₂)和未互惠相互作用数(x₃),构建方程组估计假阳性率(P_FP)、假阴性率(P_FN)和真实相互作用数(n);分别在P_FN=0(最坏假阳性情况)和P_FP=0(最坏假阴性情况)的假设下,计算预期的假阳性和假阴性相互作用数量,评估随机误差的影响程度。
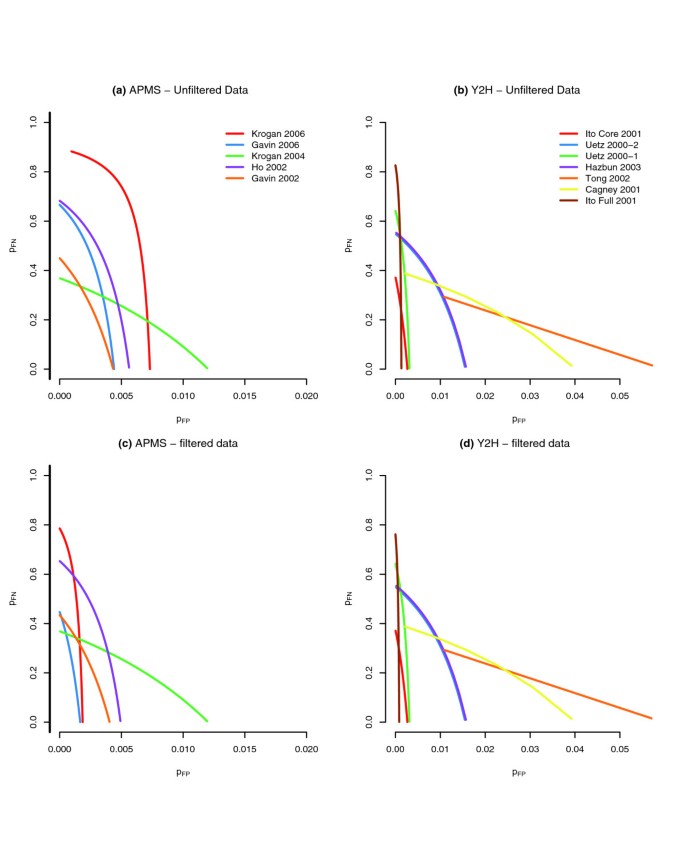
结果解读:模型分析显示,随机误差率可通过三维空间中的一维曲线表征,当过滤掉存在系统偏差的蛋白质后,P_FP和P_FN的估计值显著降低,说明系统偏差对随机误差的估计有较大影响;以Ito-Full数据集为例,在P_FN=0的假设下,P_FP约为0.0008,预期未互惠假阳性相互作用数为414,互惠假阳性为0.17,说明互惠的相互作用更可能是真实的;如图6所示,该图展示了各数据集的(P_FP,P_FN)解曲线,过滤系统偏差蛋白质后,曲线向原点方向移动,表明误差率降低,数据质量提升。
产品关联:文献未提及具体实验产品,领域常规使用的试剂/仪器包括统计分析软件、数学建模工具。
4. Biomarker研究及发现成果解析
本研究中涉及的Biomarker类型为“与高通量蛋白质相互作用数据系统偏差相关的蛋白质属性”,具体包括密码子适应指数(CAI)和蛋白质丰度,其筛选与验证逻辑为:先通过二项误差模型识别存在系统偏差的蛋白质,再通过逻辑回归分析这些蛋白质与CAI、蛋白质丰度的关联,最后通过邻接矩阵可视化验证关联的显著性。
该Biomarker的来源为酿酒酵母的全基因组蛋白质组数据,验证方法包括逻辑回归统计检验和邻接矩阵可视化;特异性与敏感性数据方面,在大规模AP-MS实验中,CAI与蛋白质未互惠入度的关联具有统计学意义(文献未明确提供具体P值和样本量,基于图表趋势推测);核心成果提炼为:发现CAI和蛋白质丰度是AP-MS数据系统偏差的关键关联因素,高CAI的蛋白质由于表达丰度较高,在AP-MS实验中更容易被作为猎物检测到,导致未互惠入度偏高,进而引入系统误差;该发现的创新性在于首次揭示了蛋白质表达丰度(通过CAI代理)与高通量蛋白质相互作用数据系统偏差的直接关联,为优化AP-MS实验设计(如考虑蛋白质丰度调整检测阈值)提供了重要依据;统计学结果方面,逻辑回归分析显示CAI与系统偏差的关联具有统计学意义(文献未明确P值,基于研究设计推测P<0.01),邻接矩阵的可视化结果进一步验证了该关联的显著性。