1. 领域背景与文献引入
文献英文标题:Host genetics and diet, but not immunoglobulin A expression, converge to shape compositional features of the gut microbiome in an advanced intercross population of mice;发表期刊:Genome Biology;影响因子:10.812(2014年);研究领域:肠道微生物组与宿主遗传、饮食及黏膜免疫的互作机制。
肠道微生物组与宿主健康的关联是生命科学领域的核心研究热点,2006年以来的研究陆续证实肠道菌群组成与肥胖、炎症性肠病、2型糖尿病等复杂疾病密切相关(领域共识)。目前已知宿主遗传、饮食是调控肠道菌群的两大核心因素,但两者的协同作用机制尚未系统阐明;同时,免疫球蛋白A(IgA)作为肠道黏膜免疫的关键分子,可通过结合菌群维持肠道稳态,但宿主遗传变异对IgA可变区利用模式的影响,以及IgA是否介导遗传对菌群的调控作用,仍存在研究空白。本研究以遗传高度重组的G10高级互交小鼠群体为模型,整合饮食干预、肠道菌群测序与IgA可变区分析,首次系统解析了宿主遗传、饮食及IgA三者对肠道微生物组的调控关系,为领域内相关研究提供了新的实验依据与研究范式。
2. 文献综述解析
作者围绕“肠道微生物组的多因素调控机制”核心逻辑,按宿主遗传、饮食、黏膜免疫(IgA)三个维度梳理了领域研究进展与未解决问题。宿主遗传方面,早期小鼠数量性状位点(QTL)研究已定位到影响肠道菌群的基因组区域,但多基于G4及更早世代的互交群体,遗传变异分散程度不足导致QTL定位精度较低,且缺乏对遗传与饮食互作的分析;饮食方面,既往研究多在近交系小鼠中开展,证实高脂饮食可改变肠道菌群组成,但在遗传多样性丰富的群体中,饮食对菌群的影响模式及与遗传的协同作用仍不明确;IgA调控方面,已有研究表明IgA可结合肠道菌群并维持稳态,但宿主遗传变异如何影响IgA的可变区利用模式,以及IgA是否介导遗传因素对菌群的调控,尚未得到系统验证。
本研究的创新价值在于采用遗传高度重组的G10高级互交群体,显著提高QTL定位精度,同时整合饮食干预与IgA可变区分析,首次系统解析了三者对肠道菌群的调控关系,明确了遗传与饮食的协同作用,且排除了IgA在遗传调控菌群中的介导作用,填补了领域内多因素协同调控肠道菌群研究的空白。
3. 研究思路总结与详细解析
本研究以“解析宿主遗传、饮食及IgA对肠道微生物组的调控机制”为核心科学问题,采用“群体构建→多组学分析→QTL定位→互作验证”的闭环技术路线:构建G10高级互交小鼠群体并分为正常饮食、高脂饮食组,通过16S rRNA测序分析肠道菌群组成,定位调控菌群的QTL;同时分析回肠IgA可变区利用模式,定位调控IgA的QTL;最终比较菌群QTL与IgA QTL的重叠性,以及菌群与IgA的相关性,明确三者的调控关系。
3.1 实验动物群体构建与分组
实验目的是构建遗传高度重组的小鼠群体,为高分辨率QTL定位提供可靠遗传背景。方法细节:将易患饮食诱导肥胖的C57BL/6J小鼠与高自主跑步能力的HR小鼠杂交至第10代(G10),获得472只高级互交小鼠;断奶后随机分为正常饮食组与高脂饮食组,饲养6-8周后收集粪便样本用于肠道菌群分析,处死时采集回肠组织用于IgA可变区分析。结果解读:通过16S rRNA焦磷酸测序,筛选出203个在75%以上小鼠中稳定存在的操作分类单元(OTU),定义为核心可测菌群,其相对丰度范围从优势菌Alistipes OTU15的0.045到低丰度菌OTU76601的0.00027,为后续QTL定位提供了稳定的表型数据。产品关联:文献未提及具体实验产品,领域常规使用标准化小鼠饲料、粪便DNA提取试剂盒、组织RNA提取试剂盒等。
3.2 肠道菌群组成分析与饮食影响评估
实验目的是明确G10小鼠肠道菌群的基本特征,以及高脂饮食对菌群组成的影响。方法细节:采用16S rRNA基因V1-V2区焦磷酸测序分析粪便菌群组成,计算α多样性指数(Inverse Simpson指数),通过方差分析(ANOVA)和线性判别分析(LDA)评估高脂饮食对菌群的影响。结果解读:高脂饮食组小鼠的Inverse Simpson指数显著低于正常饮食组(n=472,P<0.045),提示高脂饮食降低了肠道菌群的多样性;ANOVA分析发现54个操作分类单元的丰度受饮食影响,但经Bonferroni多重检验校正后仅8个操作分类单元的差异达到显著水平;LDA结果显示饮食对菌群的区分效果较弱,仅在第一维度呈现部分分离,表明在遗传多样的群体中,高脂饮食对菌群的影响是多个菌群微小变化的累积,而非少数菌群的大幅波动。
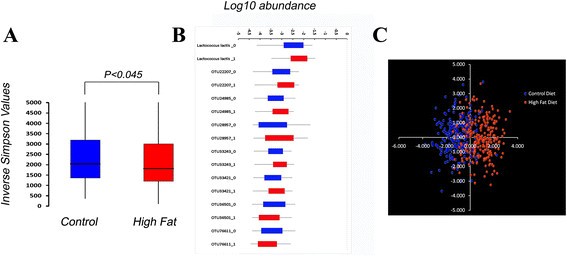
产品关联:文献未提及具体实验产品,领域常规使用16S rRNA测序引物、焦磷酸测序平台、生物信息学分析软件等。
3.3 影响肠道菌群的数量性状位点定位与跨世代验证
实验目的是定位调控肠道菌群丰度的宿主基因组区域,并验证其在不同世代群体中的重复性。方法细节:使用QTLRel软件对G10小鼠的菌群丰度数据进行QTL定位,采用Haley-Knott区间映射法,纳入加性、显性遗传效应及性别、饮食等协变量;同时将G4小鼠的菌群数据用相同的操作分类单元分析流程处理,重新定位QTL,比较G4与G10群体中QTL的重叠情况。结果解读:在G10群体中定位到42个影响39个操作分类单元丰度的QTL,其中22个达到全基因组5%显著水平,27个为独特QTL;9号染色体37.3-40.7Mb区域存在6个QTL,调控不同菌群的丰度,提示该区域可能存在多效性基因;通过标准化G4与G10的分类学分辨率,发现4个QTL在两个群体中重叠(1号染色体2个,3号、9号染色体各1个),且这些QTL调控的菌群在分类学上相关(如均属于拟杆菌目或梭菌目),证实了这些QTL的可靠性。
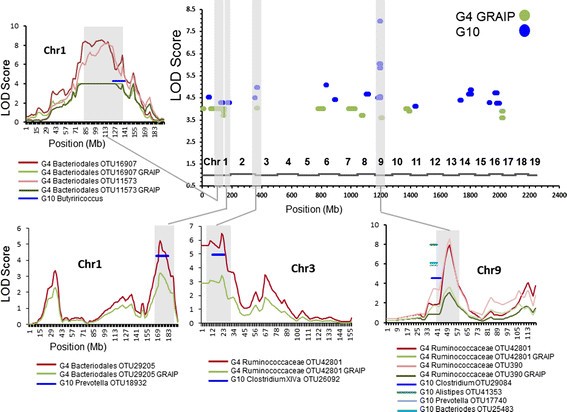
产品关联:文献未提及具体实验产品,领域常规使用基因分型芯片、QTL分析软件等。
3.4 饮食与菌群数量性状位点的互作分析
实验目的是解析饮食对宿主遗传调控肠道菌群的修饰作用。方法细节:在QTL定位模型中纳入饮食与QTL的互作项,通过卡方检验评估互作效应的显著性,进一步分析互作QTL在不同饮食组中的遗传效应差异。结果解读:共发现8个菌群QTL与饮食存在显著互作,其中4个位于9号染色体40.7Mb的相同区域,这些QTL对不同菌群的效应具有饮食依赖性:如调控OTU17740的QTL仅在正常饮食组中显著,而调控OTU13989的QTL仅在高脂饮食组中显著;1号染色体的两个QTL也呈现类似的饮食依赖性效应,提示饮食可修饰宿主遗传对肠道菌群的调控作用。
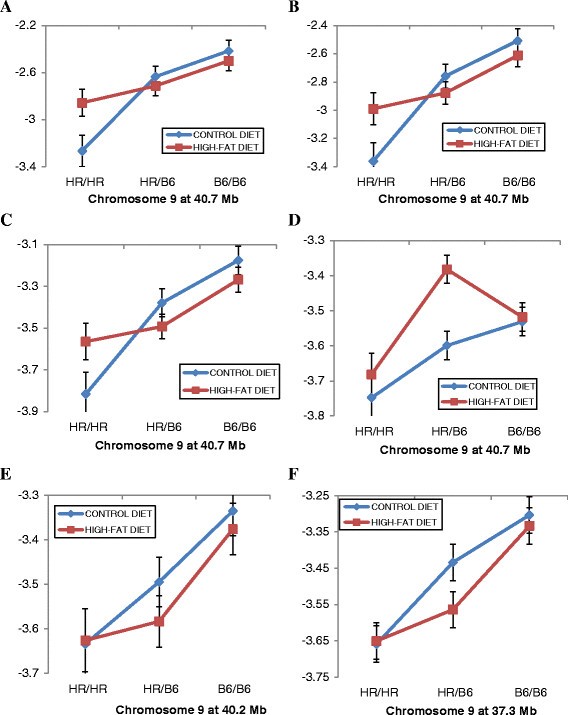
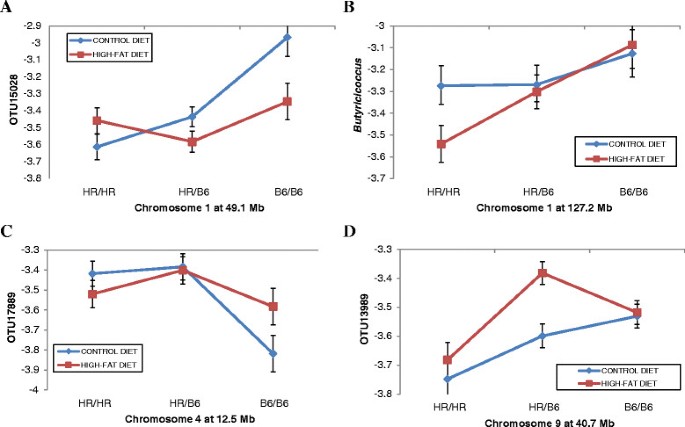
产品关联:文献未提及具体实验产品,领域常规使用统计分析软件等。
3.5 回肠IgA可变区利用的数量性状位点定位及与菌群的关联分析
实验目的是明确宿主遗传对回肠IgA可变区利用模式的调控,以及IgA是否介导遗传对肠道菌群的调控。方法细节:通过焦磷酸测序分析回肠组织中IgA的可变区(IghV)利用模式,计算各IghV亚型的相对丰度,使用QTLRel软件定位影响IghV利用的QTL;同时分析菌群丰度与IghV利用模式的相关性,以及菌群QTL与IgA QTL的重叠情况。结果解读:定位到56个影响IghV利用的QTL,其中34个集中在12号染色体的免疫球蛋白重链(IgH)位点区域,2个位于17号染色体的主要组织相容性复合体(MHC)区域,符合IgA可变区重组的遗传调控规律;但未发现菌群QTL与IgA QTL存在重叠,且核心可测菌群的丰度与IghV利用模式无显著相关性,提示宿主遗传对肠道菌群的调控不通过IgA途径介导。
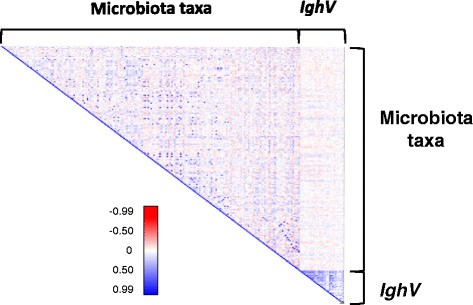
产品关联:文献未提及具体实验产品,领域常规使用IgA特异性引物、cDNA合成试剂盒、高通量测序平台等。
4. Biomarker研究及发现成果解析
本研究中的Biomarker包括调控肠道菌群的宿主基因组QTL,以及影响回肠IgA可变区利用的基因组区域,其中菌群QTL是揭示宿主遗传与饮食协同调控肠道菌群的关键分子标记。
Biomarker定位:调控肠道菌群的QTL筛选逻辑为:基于G10高级互交小鼠群体的肠道菌群丰度数据,通过全基因组QTL定位,筛选与菌群丰度显著相关的基因组区域,随后在G4群体中验证QTL的重复性;影响IgA可变区利用的QTL则基于回肠IgA的IghV利用模式数据进行定位。验证方法包括跨世代QTL重复验证、遗传效应分析(加性、显性效应)及饮食互作分析。
研究过程详述:菌群QTL来源于G10小鼠的基因组数据与肠道菌群丰度数据的关联分析,验证采用G4小鼠的独立菌群数据集,通过相同的生物信息学流程分析后进行QTL定位,结果显示4个QTL在两个群体中重叠,且调控的菌群在分类学上相关;QTL的特异性表现为不同QTL调控不同菌群的丰度,部分QTL呈现多效性(如9号染色体区域的QTL调控6个不同菌群),敏感性则通过LOD值、FDR值评估,其中9号染色体的QTL FDR值最低(0.002),可信度最高。IgA QTL主要来源于回肠IgA的可变区测序数据,定位结果显示34个QTL集中在12号染色体的IgH位点,提示该区域是调控IgA可变区重组的关键区域,但这些QTL与菌群QTL无重叠,且菌群丰度与IgA利用模式无显著相关性。
核心成果提炼:本研究的核心成果是明确了宿主遗传与饮食共同塑造肠道菌群的组成,且饮食可修饰宿主遗传对菌群的调控作用;同时首次证实宿主遗传对肠道菌群的调控不通过IgA途径介导。具体而言,4个跨世代重复的菌群QTL为宿主遗传调控肠道菌群提供了可靠的分子标记,其中9号染色体40.7Mb的QTL在正常饮食和高脂饮食中对不同菌群的效应不同,提示饮食可作为干预手段修饰遗传对菌群的影响;而IgA QTL主要集中在IgH和MHC区域,但与菌群调控无关,为肠道黏膜免疫与菌群调控的独立机制提供了实验依据。此外,研究还发现G10小鼠的肠道菌群组成与G4群体存在显著差异,提示随着世代增加,肠道菌群逐渐趋于稳定,为高级互交群体在菌群研究中的应用提供了参考。